



摘要:计算机辅助药物设计(Computcr-Aidcd Drug Dcsign,CADD)已成为当今药物研发不可或缺的一部分。采用传统的方法预测小分子结合自由能有一定的局限性,由于传统方法默认了小分子的结构信息与结合能之间是线性的关系,而对于结构化学信息二者之间并不是线性关系的情况,其预测结果并不是十分准确。随着深度学习技术的发展,我们可以通过神经网络对线性问题或者是非线性问题进行建模,以在小分子的结构信息与其结合能之间建立线性或者非线性的联系,使其预测的结果准确率有所提高。
关键词:深度学习;神经网络;小分子结合能预测;计算机辅助药物设计
中图法分类号:TP391 文献标识码:A
1 引言
计算机辅助药物设计已成为现代药物研发的重要手段,其在药物分子设计、活性预测、药效优化和副作用评估等方面的应用,将为药物研发和临床应用做出重要的贡献[1] 。这种方法的引入不仅可以辅助研发药物,甚至成为推动或决定药物研发成败的主要因素,这种方法改变了以往通过大量实验筛选进行药物研发的传统模式。结合神经网络与计算机技术进行药物研发已经越来越普遍,北京大学、中国科学院上海药物研究所、中国科学院长春应用化学研究所等高校和科研单位将人工神经网络法与分子模拟研究相结合[2~3] ,并将其直接用于指导实际的药物合成,取得了很好的研究成果。
2 相关工作
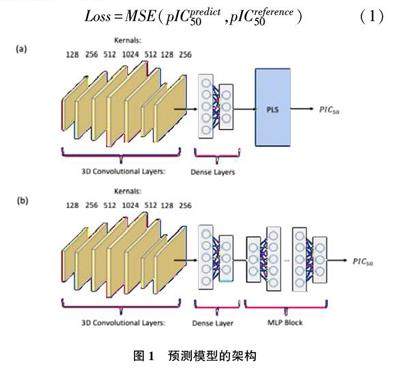
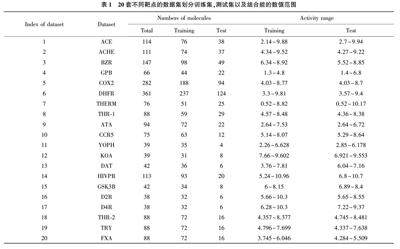
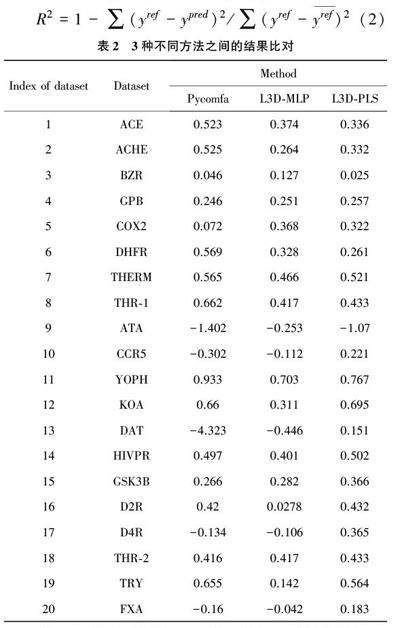
本文主要研究预测小分子的结合能,在传统的预测小分子结合能的方法基础上加入深度学习的一些方法,通过卷积神经网络提取特征,使预测小分子结合能的准确率有所提高。在此研究过程中,使用了三维定量构效关系(3D⁃QSAR)的方法,所谓三维定量构效关系是引入了分子三维结构信息并结合物理化学中常用经验方法的数学方法[4~6] 。在此基础上,利用卷积神经网络提取小分子的空间特征,小分子的空间特征主要包括小分子中原子的类型、原子的三维坐标,通过获取这些空间信息进行特征提取并找到结合能与其之间的联系,从而有利于提高预测小分子结合能的准确率。


